




200
评论
密码过期或已经不安全,请修改密码
修改密码

 添加医院
添加医院

壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




先天性甲状腺功能减退症 (congenital hypothyroidism,CH),简称先天性甲减,是指由于甲状腺激素产生不足或其受体缺陷所致的先天性疾病,可分为原发性和继发性两种。在全球范围内,先天性甲减总发病率约为1/4000~1/2000[1],且存在地域和种族差异,若不及时治疗,可引起智力障碍及生长发育落后。该病大多数病例为散发,但具体致病机制尚不清楚。近年来,随着基因芯片、全外显子测序、全基因组测序等遗传学方法的应用,越来越多与CH相关的基因被报道,为我们明确CH发病机制和病因诊断打开了一扇大门。由于CH的遗传复杂性,注定其基因诊断之路存在多重挑战。那么在临床工作中,哪些患者需要开展基因检测呢?为了回答这个问题,我们必需首先了解CH的相关遗传机制。

罗小平 教授
华中科技大学同济医学院儿科学系主任
附属同济医院儿科学系主任
同济儿童医院院长
二级教授、主任医师,医学博士、博士生导师
国家杰出青年科学基金获得者
中华医学会儿科学分会副主任委员
内分泌遗传代谢学组名誉组长
中国医师协会儿科分会常务委员
青春期医学专业委员会副主任委员
中国医疗保健国际交流促进会妇儿专委会副主任委员
儿科专委会副主任委员
中国医院协会罕见病专委会副主委
中国医药教育协会儿科副主委
中国研究型医院学会儿科副主委
世界中医药学会联合会优生优育专业委员会常务副会长
湖北省儿科学会主任委员
湖北省围产医学会名誉主任委员
亚太儿童内分泌学会前主席
亚洲遗传代谢病学会理事
生长激素研究学会理事
国内外40余种杂志主编/副主编/编委
主编参编教材专著60余部,发表论文530余篇
国家科技进步奖二等奖、湖北省科技进步奖一等奖、
自然科学一等奖、教学成果一等奖等
首届中国儿科医师奖、首届国之名医、
出生缺陷防控杰出贡献奖、宋庆龄儿科医学奖等
卫健委有突出贡献中青年专家、享受政府津贴专家、
新世纪百千万人才工程国家级人选

叶枫 教授
华中科技大学同济医学院附属同济医院儿科学系
博士、副主任医师、副教授、硕士生导师
中华医学会儿科学分会临床营养学组委员
中华医学会儿科学分会临床药理学组青年委员
中国医师协会青春期健康与医学专业委员会委员
主持国家级、省级等基金项目六项,发表论文20余篇
获国际甲状腺会议(ITC)“青年研究奖”
获中华医学会内分泌学分会默克雪兰诺甲状腺奖
多次获华中科技大学教学竞赛一等奖
擅长:儿科遗传代谢内分泌疾病的诊断和治疗,
如:儿童身材矮小、性早熟、糖尿病和甲状腺疾病等
原发性甲减相关基因
原发性CH通常是由甲状腺发育不良(thyroid dysgenesis,TD)或甲状腺激素生成障碍(dyshormonogenesis,DH)引起。其中CHTD 约占 CH 的 65%,是永久性CH最常见原因,目前在不到5%的患者中发现了致病基因。其余 35% 为CHDH病例,在超过50%的病例中,可以在分子水平找到病因[2, 3]。甲状腺发育任何阶段的异常(如甲状腺增殖、迁移、生长、组织形成、分化和存活),或甲状腺激素生物合成过程中任何环节的缺陷,均可能导致不同程度的甲状腺功能减退症[4]。
CHTD包括甲状腺缺失(35–40%)、甲状腺异位(30–45%,最常见于舌下)以及发育不全或单叶(5%)[4, 5],通常为散发性,但有2%的病例为家族性。大多数甲状腺发育不全及其相关疾病为常染色体显性遗传,虽然只有不到5%的病例发现了致病基因,但这些已知的遗传病因对了解甲状腺个体发育具有重要意义。例如,TSHR[6]和转录因子PAX8、NKX2-1和FOXE1等均在发育的甲状腺中表达,这些基因中的任何一个被破坏都可能导致甲状腺发育异常,有些基因还参与其他组织的发育,从而形成相应的综合征[7]。目前发现的与甲状腺发育不良相关的主要基因及其特点见表1[2, 4, 8]。值得注意的是,有些在动物实验中发现致病的基因,在人体中的致病性尚不明确[9]。还有些基因存在高度的异质性,这些均增加了我们对基因报告解读的难度。而同卵双胞胎中甲状腺发育的高度不一致性也进一步说明CHTD病因复杂,无法单独用遗传因素解释[10]。

甲状腺激素生成障碍相关基因
甲状腺激素生成障碍通常是由参与甲状腺激素合成的基因缺陷引起的,例如TSH受体信号转导或碘化物转运或活化,TH合成所必需的任何蛋白质的缺陷均有可能导致CHDH。在这些患者中,甲状腺的发育和分化是正常的,但甲状腺细胞中TH合成的关键环节发生了异常,甲状腺B超通常显示甲状腺正常或者肿大。与CHTD不同,大多数CHDH以常染色体隐性遗传模式遗传,通常没有相关的畸形。目前发现的与甲状腺激素生成障碍相关的基因主要有GNAS、SLC5A5、SLC26A4、TPO、TG、IYD、DUOXA2、DUOX1、DUOX2和SLC26A7等,致病特点见表2[2, 4, 8, 11, 12]。CHTD和CHDH相关的基因之间存在一些表型重叠,很大一部分 CH 病例可能是由 2 个或多个 CH 相关基因突变之间的相互作用引起的。

继发性甲减相关基因
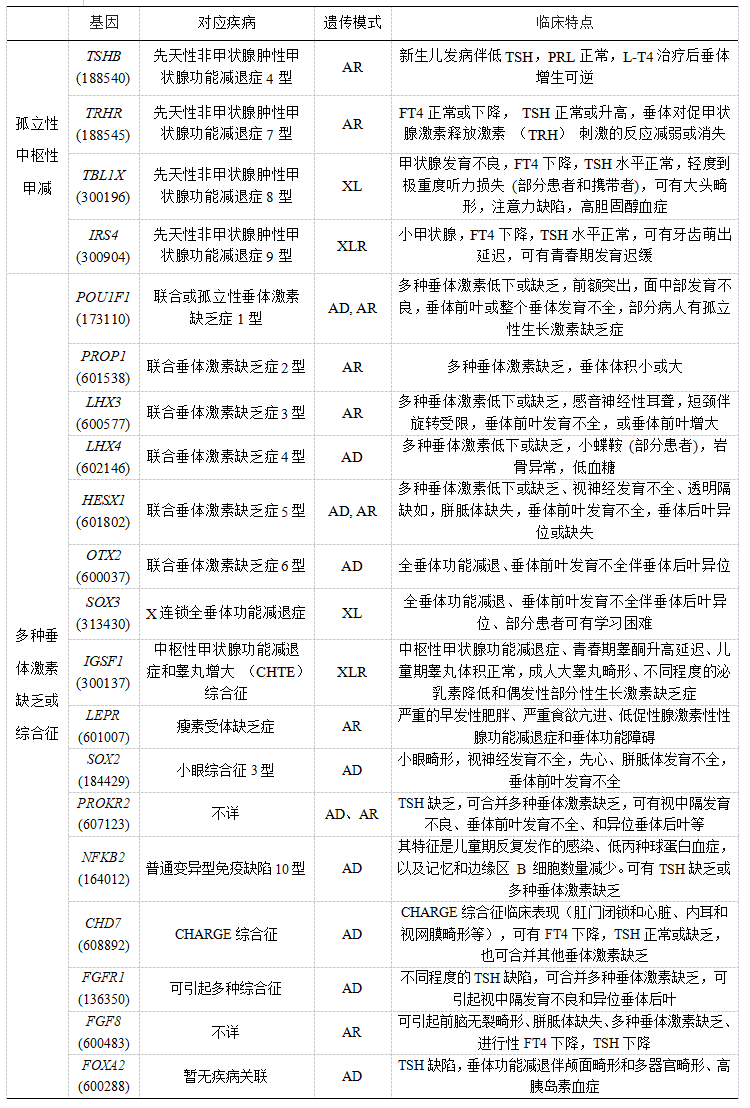
继发性甲减,又称中枢性甲减,比较罕见,发病率约为1/25 000~1/16 000,是由下丘脑或垂体的异常发育或损害TRH或TSH功能的遗传改变引起的。下丘脑和/或垂体发育或结构异常可导致多种垂体激素缺乏。而TRH或TSH信号转导的特定缺陷可导致孤立性中枢性CH。目前发现的与孤立性中枢性CH相关的基因主要有TSHB、TRHR、TBL1X、IRS4,与多种垂体激素缺乏相关的基因主要有IGSF1、PROP1、POU1F1、HESX1、SOX3、OTX2、LHX3、LHX4,还有一些基因可引起各种综合征,包括LEPR、SOX2、PROKR2、NFKB2、CHD7、FGFR1、FGF8、FOXA2等,这些基因的临床特点具体见表3[2, 4, 8, 12, 13]。
CH的遗传复杂性
虽然CH潜在的分子和遗传机制已经被阐明,但很多病例依然找不到确切原因,尤其是在CHTD中。最近一项成人CH患者的研究发现甲状腺肿组、正常腺体组和发育不全组遗传缺陷患者的比例分别为89%、36%和9%[14]。除了罕见的单基因形式外,CH可能更频繁地出现在散发病例中。同卵双胞胎之间的高度不一致 (92%) 和性别差异以及CHTD女性患病率较高等,均表明CH是复杂遗传性疾病,受环境和遗传因素的多重影响。Luca Persani团队在177名CH患者队列中发现了11个致病基因,提出散发性CH可能是由多个不同CH基因缺陷共同引起[15, 16]。这种寡基因模型可以解释CH家族病例中存在的外显不全,但却无法解释同卵双胞胎(理论上具有完全相同的基因型)发病率高度不一致现象,因此也有学者提出CH可能与表观遗传学修饰或者体细胞突变等有关[9, 10]。甚至有学者推测遗传学不是导致散发性CHTD 的主要因素[17]。
CH的遗传复杂性,以及高额的基因检测费用,导致临床医生对CH患者是否进行基因检测难以决断。纵观目前的研究结果,我们建议:①对所有具有综合征特点的甲减患者进行基因检测;②对CHDH、中枢性甲减患者以及有家族史的CHTD患者,因基因突变致病的风险相对较高,可考虑行基因检测明确病因;③对于散发的CHTD患者,因目前的遗传机制尚不明确,不建议常规行基因检测。目前的基因检测相关技术包括:比较基因组杂交技术、基因panel检测、全外显子组测序等。为了提高检测效率,在送检基因时,需要详细描述患者临床表型,包括甲状腺功能、甲状腺形态、甲状腺外组织的表现等等。当试图阐明病因时,其他遗传和环境因素也需要考虑,包括调控区,内含子突变和拷贝数变异等。未来,其他可能的机制,例如表观遗传学修饰等,也需要我们进一步探索。
参考文献:
[1]ROSE S R, WASSNER A J, WINTERGERST K A, et al. Congenital Hypothyroidism: Screening and Management[J]. Pediatrics, 2023,151(1).
[2]STOUPA A, KARIYAWASAM D, POLAK M, et al. Genetics of congenital hypothyroidism: Modern concepts[J]. Pediatr Investig, 2022,6(2): 123-134.
[3]ACAR S, GURSOY S, ARSLAN G, et al. Screening of 23 candidate genes by next-generation sequencing of patients with permanent congenital hypothyroidism: novel variants in TG, TSHR, DUOX2, FOXE1, and SLC26A7[J]. J Endocrinol Invest, 2022,45(4): 773-786.
[4]KOSTOPOULOU E, MILIORDOS K, SPILIOTIS B. Genetics of primary congenital hypothyroidism-a review[J]. Hormones (Athens), 2021,20(2): 225-236.
[5]BARRY Y, BONALDI C, GOULET V, et al. Increased incidence of congenital hypothyroidism in France from 1982 to 2012: a nationwide multicenter analysis[J]. Ann Epidemiol, 2016,26(2): 100-105.
[6]De FELICE M, Di LAURO R. Thyroid development and its disorders: genetics and molecular mechanisms[J]. Endocr Rev, 2004,25(5): 722-746.
[7]CHERELLA C E, WASSNER A J. Congenital hypothyroidism: insights into pathogenesis and treatment[J]. Int J Pediatr Endocrinol, 2017,2017: 11.
[8]van TROTSENBURG P, STOUPA A, LEGER J, et al. Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update-An ENDO-European Reference Network Initiative Endorsed by the European Society for Pediatric Endocrinology and the European Society for Endocrinology[J]. Thyroid, 2021,31(3): 387-419.
[9]VASSART G, DUMONT J E. Thyroid dysgenesis: multigenic or epigenetic ... or both?[J]. Endocrinology, 2005,146(12): 5035-5037.
[10]PERRY R, HEINRICHS C, BOURDOUX P, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology[J]. J Clin Endocrinol Metab, 2002,87(9): 4072-4077.
[11]ACAR S, GURSOY S, ARSLAN G, et al. Screening of 23 candidate genes by next-generation sequencing of patients with permanent congenital hypothyroidism: novel variants in TG, TSHR, DUOX2, FOXE1, and SLC26A7[J]. J Endocrinol Invest, 2022,45(4): 773-786.
[12]PETERS C, van TROTSENBURG A, SCHOENMAKERS N. DIAGNOSIS OF ENDOCRINE DISEASE: Congenital hypothyroidism: update and perspectives[J]. Eur J Endocrinol, 2018,179(6): R297-R317.
[13]MCCABE M J, GASTON-MASSUET C, GREGORY L C, et al. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia[J]. J Clin Endocrinol Metab, 2013,98(3): E547-E557.
[14]SUGISAWA C, NARUMI S, TANASE-NAKAO K, et al. Adult Thyroid Outcomes of Congenital Hypothyroidism[J]. Thyroid, 2023,33(5): 556-565.
[15]de FILIPPIS T, GELMINI G, PARABOSCHI E, et al. A frequent oligogenic involvement in congenital hypothyroidism[J]. Hum Mol Genet, 2017,26(13): 2507-2514.
[16]OLIVER-PETIT I, EDOUARD T, JACQUES V, et al. Next-Generation Sequencing Analysis Reveals Frequent Familial Origin and Oligogenism in Congenital Hypothyroidism With Dyshormonogenesis[J]. Front Endocrinol (Lausanne), 2021,12: 657913.
[17]LARRIVEE-VANIER S, JEAN-LOUIS M, MAGNE F, et al. Whole-Exome Sequencing in Congenital Hypothyroidism Due to Thyroid Dysgenesis[J]. Thyroid, 2022,32(5): 486-495.
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017